A Network-Based Gene Weighting Approach for Pathway Analysis
Source:
Time: 2011-09-15
To infer biological pathways with altered activities is one of the frequently encountered tasks in genome-wide studies, and many algorithms have already been developed, with a predominant emphasis on gene expression microarray experiments. Most algorithms designed in line have been based on the reduction of complex signaling pathways into gene sets, which is convenient to methodological development. However, it may be beneficial to take into account as well the inter-gene functional associations when testing pathway significance. A recent collaboration work between JI Hongbin at the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, CAS and TIAN Weidong at the School of Life Sciences, Institute of Biostatistics, Fudan University provided a new pathway analysis framework by taking a gene functional association network-based gene weighting approach.
With the notion that genes in a given pathway can have a different relative importance, which is termed “constitutive nonequivalence” in the work, FANG Zhaoyuan, under the supervision of JI Hongbin and TIAN Weidong, designed a network-based method to calculate gene weights, which has been shown to fit biological knowledge as well as robust against a certain level of random perturbations in the network. The authors proposed that the gene weights could be integrated with many previous pathway analysis algorithms, two of which (MeanAbs and GSEA) were taken to illustrate this point. With testing on various datasets, they demonstrated a unique advantage of the weighted approach for pathways consisted of heterogeneous genes or gene groups, which have different association strength to the pathway they belonging to. The authors further targeted the “multiple-subunit protein” issue which is often ignored. Multiple-subunit proteins have each subunit encoded by a different gene following the nomenclature tradition, which resulted a natural “overweighting” for such proteins when compared with single gene-encoding proteins. To correct for such bias, the authors seamlessly adapted their gene-level gene weighting method to protein-level. Finally, a better reproducibility was shown for the weighted approach with three independent breast cancer datasets.
This work entitled “A Network-Based Gene Weighting Approach for Pathway Analysis” was published in Cell Research online in advance on Sep 6, 2011.
This study was supported by the grants from the National Basic Research Program of China, the National Natural Science Foundation of China, the Chinese Academy of Sciences, and the Science and Technology Commission of Shanghai Municipality.
AUTHOR CONTACT:
JI Hongbin
Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China
Phone: 86-21-54921101; E-mail: hbji@sibs.ac.cn
TIAN Weidong
School of Life Sciences, Institute of Biostatistics, Fudan University, Shanghai, China
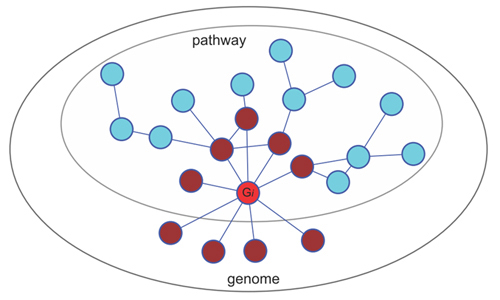
An illustration of the gene weighting algorithm. A gene functional association network is required to provide inter-gene association linkages. For any gene (red) in a given pathway of genes, its functional associated genes (brown) are distributed within (Xi) and outside (Mi-Xi) the pathway. A hypergeometric distribution model can be used to estimate the specific associations between and , which reflects ’s contribution to the pathway.
(Image provided by Dr. JI Hongbin)
 Appendix:
Appendix: