β-Arrestin1, a New Way to Tackle Alzheimer’s Disease
Source:
Time: 2012-12-13
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the most common form of dementia, accounting for 50 to 70 percent of worldwide dementia cases. According to the World Alzheimer Report 2010, the worldwide costs of dementia (US$604 billion in 2010) amount to more than 1% of global GDP. If dementia care were a country, it would be the world’s 18th largest economy. Despite tens of thousands of scientific articles about neurodegeneration, we are not closer to a cure of the disease. Now researchers from Chinese Academy of Sciences unraveled a novel mechanism regulating the amyloidogenic processing of APP and provided a potential therapeutic strategy against AD.
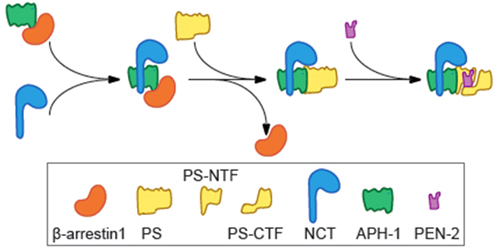
The major sporadic AD cases exhibit onset in their seventies or even later, whereas the fewer, mutation linked, familial AD cases exhibit onset typically during their fifties. Most cases of AD suffer from neuron loss, synaptic dysfunction, cognitive impairment and inability to function independently. Decades of researches have pathologically characterized AD by two proteinaceous aggregates: amyloid plaques composed of amyloid-β (Aβ) peptides and neurofibrillary tangles consisting of the hyperphosphorylated microtubule-associated protein tau. The Aβ peptides are products of sequential processing of amyloid-β precursor protein (APP) by BACE (β-amyloid cleaving enzyme) and γ-secretase complex, comprising at least presenilin (PS), nicastrin (NCT), anterior pharynx-defective phenotype 1 (APH-1) and presenilin enhancer 2 (PEN-2).
Dr. PEI Gang, Dr. ZHAO Jian and their colleagues from Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences found that a multifunctional protein, β-arrestin1, facilitated the NCT/APH1 precomplex and mature γ-secretase complex formation through its functional interaction with APH-1. Deficiency of β-arrestin1 or inhibition of binding of β-arrestin1 with APH-1 by small peptides reduced Aβ production without affecting the proteolysis activity of γ-secretase to other substrates including Notch. Genetic ablation of β-arrestin1 diminished Aβ pathology and behavioral deficits in transgenic AD model mice. Moreover, in sporadic AD patients and transgenic AD mice brains, the expression of β-arrestin1 was upregulated and correlated well with neuropathological severity and senile Aβ plaques, indicating a physical relevance of β-arrestin1 with AD pathogenesis. This study not only identifies a regulatory mechanism underlying both γ-secretase assembly and AD pathogenesis, but indicates specific reduction of Aβ pathology can be achieved by regulation of the γ-secretase assembly.
This study has been entitled as “β-Arrestin1 regulates γ-secretase complex assembly and modulates amyloid-β pathology ” and online published on Cell Research on Dec. 4th.
AUTHOR CONTACT:
PEI Gang
Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai, China
ZHAO Jian
Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai, China
Mediation of γ-secretase complex assembly by β-Arrestin1
 Appendix:
Appendix: