On April 10, the academic journal《Nucleic Acids Research》online published the new work from En-Duo Wang’s group in Shanghai Institute of Biochemistry and Cell Biology entitled “A natural non-Watson-Crick base pair in hmtRNAThr causes structural and functional susceptibility to local mutations”.
Mitochondria are essential organelle to regulate the life cycle of eukaryotic cells. They have their own genome and protein synthesis system. Mitochondrial gene mutations are often related with various mitochondrial diseases. Human mitochondrial tRNAThr (hmtRNAThr) is encoded by mitochondrial genome and it has one gene copy. hmtRNAThr reads all the threonine codon in mitochondrial translation system and is important for mitochondrial function and homeostasis. So far, six pathogenic mutations have been reported in human mitochondrial tRNAThr which probably affect the structure and function of hmtRNAThr and then the mitochondrial protein synthesis. However, the pathogenic molecular mechanism remains unclear.
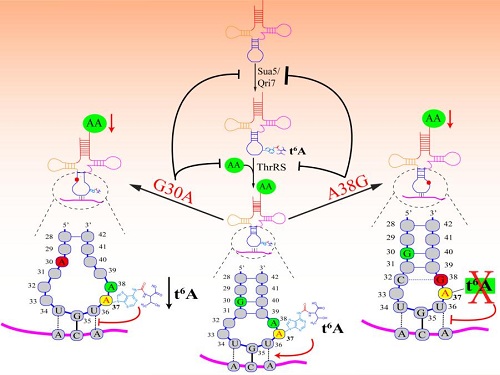
Under the guidance of Profs. En-Duo Wang and Xiao-Long Zhou, Ph. D students Yong Wang et al. focused on two pathogenic mutations G30A and A38G in the anticodon stem and loop which severely affect the structure and aminoacylation of tRNA. Because of the evolutionary gain of non-Watson-Crick base pair A29/C41, the third base pair in the anticodon stem of hmtRNAThr,which is different with other mitochondrial tRNAThr of primates like Pan troglodytes, the fourth Watson-Crick base pair G30-C40 in the anticodon stem is critical in maintaining the proper structure and function of hmtRNAThr. The pathogenic mutation G30A affected the structure, function and posttranscriptional modification of tRNA by disrupting the fourth Watson-Crick base pair G30-C40 in the anticodon stem. The pathogenic mutation A38G in the anticodon stem and near the anticodon resulted in decreased amino acid accepting capability and abolition of posttranscriptional modification at position 37 which is essential for maintaining the translational fidelity. The data suggested that the pathogenic mutations G30A and A38G reduced the substrates for protein biosynthesis and translational fidelity. In summary, the results revealed the pathogenic mutations G30A and A38G led to mitochondrial diseases by reducing the efficiency and fidelity of mitochondrial protein synthesis and it is important for understanding the pathogenic mechanism of mitochondrial tRNA mutations.
This work was supported by grants from the National Key Research and Development Program of China; National Natural Science Foundation of China; Strategic Priority Research Program of the Chinese Academy of Sciences; Shanghai Rising-Star Program; Youth Innovation Promotion Association, CAS (to Xiao-Long Zhou).
 Appendix:
Appendix: