Genetics and epigenetics are inextricably linked in the pathogenesis of cancer, among which epigenetic aberrations are frequently observed and also known to cooperate with genetic alterations to drive malignant transformation. Among these aberrations, oncogenic imbalance of DNA methylation is well-recognized as a classic hallmark of cancer.
The ten-eleven translocation (TET) dioxygenases, which can facilitate DNA oxidative demethylation, are frequently mutated or misregulated in hematological malignancies. However, given the differential mutational incidence, whether and how mutated TET genes endow cells a selective growth advantage in solid tumors remain underexplored.
In a study published online in PNAS, the groups of Prof. XU Guoliang and Prof. JI Hongbin from the Center for Excellence in Molecular Cell Science (CEMCS), Shanghai Institute of Biochemistry and Cell Biology of the Chinese Academy of Science, reported that loss-of-function TET enzymes cooperated with oncogenic KRAS to induce augmented Wnt signaling through transcriptional silencing of Wnt-associated antagonizing genes arising from DNA hypermethylation, contributing to aggressive lung cancer development.
The researchers firstly identified TET mutations in 7.4% of human lung adenocarcinoma (LUAD), which frequently co-occurred with oncogenic mutations of KRAS, but not EGFR, and this co-occurrence was predictive of poor survival in LUAD patients. Furthermore, human LUAD-derived TET mutations impaired their own gene expression and catalytic activity. Correspondingly, lower TET expression was associated with worse patient survival.
To recapitulate these clinical observations, the researchers generated TET-deficient genetically engineered mouse models (GEMMs) in the setting of mutant KRAS. Inactivation of any Tet family genes strongly potentiated the development of KrasG12D-driven LUAD in mice, and this strong tumorigenic phenotype could be largely mitigated by the restoration of TET dioxygenase activity.
Mechanically, TET deficiency triggered neoplastic reprogramming of DNA methylation and gene expression in premalignant cells, featuring a particular impact on Wnt signaling. During this reprogramming, multiple canonical Wnt antagonizing genes appeared silenced expression arising from elevated DNA methylation both in mice and human LUAD, leading to the hyperactivation of Wnt signaling.
Consequently, genetic depletion of β-catenin, the transcriptional effector of Wnt signaling, substantially alleviated the malignant progression of TET-deficient LUAD, highlighting the cross talk between TET and Wnt signaling.
This study establishes the first TET-deficient GEMMs in solid tumors to faithfully recapitulate the genetic alterations observed in human LUAD, and provides genetic evidence for the mutational cooperativity between TET genes and oncogenic KRAS in the induction of malignant LUAD. Therapeutically, these findings pinpoint a vulnerability that may be exploited for TET deficiency-induced lung cancer through targeting Wnt signaling, and shed light on epigenetic interventions to correct aberrant methylation as one promising modality for lung cancer treatment.
Future studies can leverage these insights and preclinical models to explore how LUAD patients harboring TET mutations obtain therapeutic benefits from manipulating Wnt signaling pathway and epigenetic therapy.
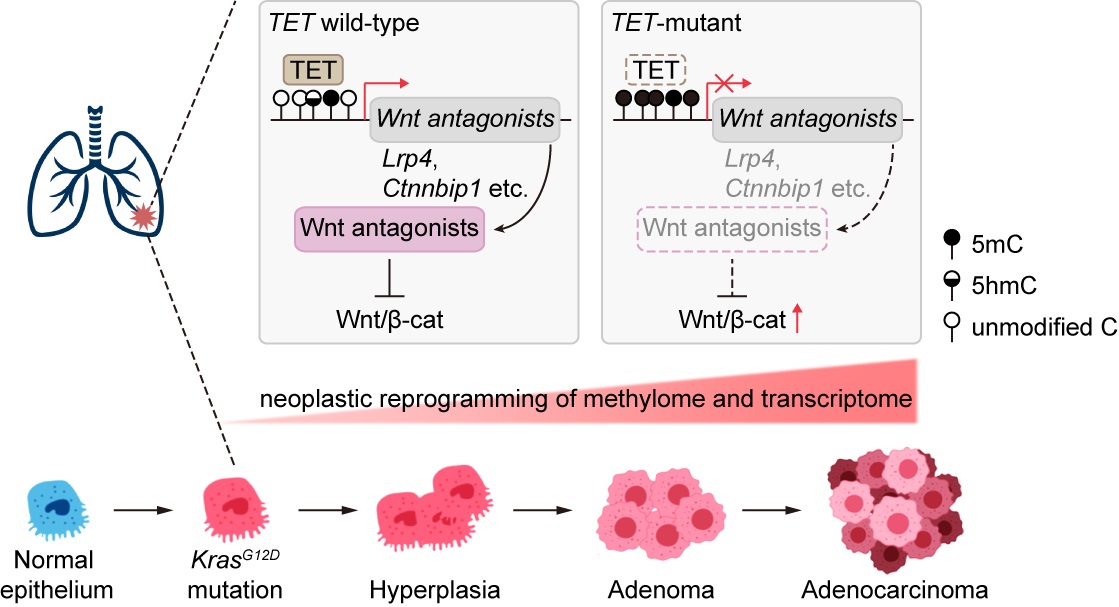
Working model illustrating the contribution of TET enzymes to antagonizing Kras-driven LUAD development (Image by CEMCS)
Contact: glxu@sibcb.ac.cn, hbji@sibcb.ac.cn
Reference: https://doi.org/10.1073/pnas.2107599119
 Appendix:
Appendix: